Research Report
A Hierarchical Inference Framework for Multi-Trait Genetics Integrating Genomic SEM, PLEIO, and Primo 
 Author
Author  Correspondence author
Correspondence author
Tree Genetics and Molecular Breeding, 2026, Vol. 16, No. 1
Received: 17 Mar., 2026 Accepted: 12 Apr., 2026 Published: 15 May, 2026
Complex traits are typically characterized by substantial genetic correlation and pleiotropy. However, conventional single-trait GWAS frameworks are limited in their ability to distinguish true shared causal effects from spurious associations arising from linkage disequilibrium, sample structure, or mediated relationships. Here, we advance multi-trait analysis from a collection of methods to a unified statistical genetic framework centered on clearly defined estimands, establishing a hierarchical inference system spanning covariance structure, locus configuration, and association patterns. Within this framework, Genomic SEM characterizes cross-trait genetic covariance and latent shared factors at the structural level; PLEIO performs joint fine-mapping within local LD structure to resolve causal configurations at the locus level; and Primo decomposes multi-trait association patterns using Bayesian mixture modeling to quantify shared and trait-specific effects at the pattern level. These approaches correspond to distinct inferential layers and collectively form a progressive evidence chain from structural reconstruction to effect decomposition. Through simulation and empirical analyses, we systematically evaluate the bias–variance trade-offs of multivariate methods under varying genetic correlation, LD complexity, and sample overlap scenarios, delineating the conditions under which multi-trait models improve power versus inflate false positives. We further emphasize a multi-evidence framework integrating local genetic correlation, joint fine-mapping, colocalization, effect direction consistency, and cross-ancestry validation to distinguish true pleiotropy from spurious signals. Building on this theoretical foundation, we propose a structured “screen–validate–apply” workflow: screening trait sets via genome-wide and local genetic correlation, resolving shared architectures using PLEIO and Primo, validating consistency through colocalization and conditional analyses, and finally expanding shared signals via Genomic SEM for downstream network and functional interpretation. This framework is broadly applicable to both crop genetics and human disease studies, providing a systematic pathway from statistical association to mechanistic insight and translational application.
1 Introduction
Genetic correlations across multiple traits are not an exception but a defining feature of complex biological systems. Across both agricultural and human contexts, phenotypes often exhibit coordinated variation. For example, yield-related traits and stress resistance in crops are shaped by shared developmental and resource allocation processes, while metabolic traits and psychiatric disorders in humans show substantial cross-trait genetic overlap. Such patterns may arise from pleiotropy, where a single genetic variant influences multiple phenotypes, but can also reflect linkage disequilibrium or mediated relationships among traits. Large-scale meta-analyses have consistently demonstrated the ubiquity of cross-trait genetic associations, highlighting that a substantial proportion of variants contribute to multiple phenotypes and underscoring the importance of characterizing shared genetic architecture.
At the statistical level, this shared architecture is typically summarized using genetic correlation (rg) or, more generally, the genetic covariance matrix (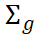 ). However, conventional single-trait GWAS are designed to estimate marginal effects for individual traits and do not directly capture cross-trait structure. As a result, signals that are weak but consistent across multiple traits may remain undetected, while post hoc integration of independent GWAS results often fails to recover the underlying covariance structure. Moreover, such approaches are susceptible to biases introduced by sample overlap, measurement error, and environmental confounding, increasing the risk of attributing apparent pleiotropy to linkage disequilibrium or indirect causal pathways. Ignoring cross-trait covariance can also distort effect estimates and their uncertainty, with downstream consequences for multi-trait prediction and selection in both breeding and precision medicine. This limitation reflects the broader evolution of statistical genetics from hypothesis-driven mapping to genome-wide association modeling, while highlighting the need for frameworks beyond single-trait GWAS (Fang and Wu, 2026).
). However, conventional single-trait GWAS are designed to estimate marginal effects for individual traits and do not directly capture cross-trait structure. As a result, signals that are weak but consistent across multiple traits may remain undetected, while post hoc integration of independent GWAS results often fails to recover the underlying covariance structure. Moreover, such approaches are susceptible to biases introduced by sample overlap, measurement error, and environmental confounding, increasing the risk of attributing apparent pleiotropy to linkage disequilibrium or indirect causal pathways. Ignoring cross-trait covariance can also distort effect estimates and their uncertainty, with downstream consequences for multi-trait prediction and selection in both breeding and precision medicine. This limitation reflects the broader evolution of statistical genetics from hypothesis-driven mapping to genome-wide association modeling, while highlighting the need for frameworks beyond single-trait GWAS (Fang and Wu, 2026).
To address these limitations, multivariate frameworks have been developed with a shift in focus from individual trait effects to the structure of genetic relationships across traits. These approaches can be broadly categorized into three complementary paradigms. First, Genomic SEM models the genetic covariance matrix at the latent variable level, enabling the decomposition of shared and trait-specific components and providing a structured representation of cross-trait relationships (Grotzinger et al., 2019). Second, methods such as PLEIO operate at the locus level, jointly modeling multi-trait effects under LD constraints to distinguish shared from trait-specific signals. Third, probabilistic approaches such as Primo treat patterns of association across traits as the primary unit of inference, estimating posterior probabilities for different configurations of shared and specific effects (Gleason et al., 2020). Despite their methodological differences, these approaches share a common objective: to characterize the statistical form of shared genetic architecture rather than to directly estimate causal effects.
It is important to note that shared association signals do not necessarily imply causal pleiotropy. Distinguishing true pleiotropy-where a single causal variant affects multiple traits-from apparent overlap driven by LD, mediation, or confounding remains a central challenge. Multivariate analyses address whether genetic signals are shared, but do not, on their own, establish whether such sharing reflects a common causal mechanism. Interpreting multivariate results as evidence of causality may therefore lead to inflated false positives. Increasingly, studies have emphasized that multivariate methods should be integrated with fine-mapping, colocalization, and causal inference approaches such as Mendelian randomization to construct a layered evidence framework that bridges shared structure and causal interpretation.
Building on this perspective, we position multivariate analysis within a broader causal inference framework in statistical genetics. Specifically, Genomic SEM, PLEIO, and Primo are treated as tools operating at the structural, locus, and pattern levels, respectively, each targeting a distinct aspect of cross-trait genetic architecture. By clarifying their roles and limitations, we propose an analytical strategy that combines structure reconstruction, locus-level resolution, and pattern decomposition, followed by downstream integration with colocalization and causal inference. This framework provides a coherent pathway from shared genetic signals to interpretable biological hypotheses, facilitating a more robust transition from association to causality in both plant and human genetics.
2 Statistical Background of Multi-trait Analysis and Pleiotropy
2.1 Estimation of genetic correlation
The starting point of multi-trait analysis lies in characterizing the dependence structure among phenotypes at the genetic level. Statistically, this structure is typically summarized by the genetic correlation (rg) or, more generally, the genetic covariance matrix (), which captures the extent to which additive genetic effects co-vary across traits. Unlike single-trait analyses that focus on marginal effects for individual phenotypes, multi-trait frameworks treat cross-trait genetic structure itself as the primary object of inference.
In practice, estimation of genetic correlation generally follows two complementary approaches: individual-level mixed model methods and summary-statistic-based regression methods. The former is exemplified by bivariate GREML, which directly decomposes genetic variance and covariance within a linear mixed model, whereas the latter is represented by cross-trait LD Score Regression (LDSC), which infers genetic covariance from the relationship between LD structure and GWAS test statistics.
At the individual level, bivariate GREML assumes that, for traits k∈{1,2}, the phenotype can be modeled as:
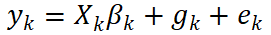
where gk denotes additive genetic effects. The joint distribution can be written as:
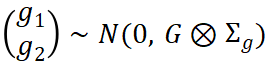
from which the genetic covariance matrix can be estimated, leading to:
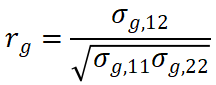
This approach provides high precision when sample size is sufficient and allows flexible modeling of covariates and extensions to complex designs such as multi-environment or multi-population settings. However, it requires access to individual-level data and is sensitive to the modeling of ancestry structure and relatedness (Zhang et al., 2015).
In contrast, cross-trait LDSC operates on summary statistics. It uses the LD score for each variant, 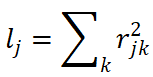 , to regress the expected product of GWAS z-scores across traits
, to regress the expected product of GWAS z-scores across traits
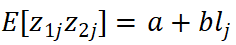
where the slope b is proportional to the genetic covariance, and the intercept a captures inflation due to sample overlap and population structure. This framework is well suited for large-scale meta-analysis without requiring individual-level data, but its validity depends on appropriate LD reference panels and model assumptions.
It is important to note that genome-wide average rg may obscure substantial heterogeneity at the regional level. Methods for local genetic correlation (e.g., HESS, LAVA) and stratified LDSC enable decomposition of shared genetic effects across genomic regions and functional annotations. Such refinement improves biological interpretability and provides more informative priors for downstream analyses, including colocalization and causal inference. In large-scale applications, transparent reporting of sample overlap, ancestry composition, and control strategies is essential to avoid misinterpreting structural confounding as shared genetic effects (Zhang et al., 2015).
2.2 Distinguishing true pleiotropy from apparent pleiotropy
A central challenge in multi-trait analysis is to distinguish genuine shared genetic effects from apparent correlations driven by confounding factors. True pleiotropy refers to a single causal variant influencing multiple traits, either directly or through mediated pathways. In contrast, apparent pleiotropy arises from linkage disequilibrium, population structure, sample overlap, or unmodeled causal relationships.
From an evidential perspective, true pleiotropy is typically characterized by consistent signals across traits at the locus level. For example, colocalization analyses may indicate a high posterior probability of a shared causal variant, credible sets may substantially overlap, and effect directions may be consistent or biologically interpretable. These signals also tend to be robust to confounding adjustment and reproducible across populations. Methods such as joint fine-mapping, Bayesian colocalization, and latent variable models (e.g., Genomic SEM) can provide complementary evidence at different levels (Zhang et al., 2015).
By contrast, apparent pleiotropy often manifests as unstable or context-dependent associations. Distinct causal variants in strong LD may produce overlapping signals that separate upon fine-mapping; sample overlap or population structure may inflate cross-trait correlations; and unmodeled mediation pathways may generate indirect associations. These patterns are typically sensitive to LD reference panels and ancestry composition and may fail to replicate across populations (Wang et al., 2024).
Consequently, inference of pleiotropy should not rely on a single analytical step but rather on a layered evaluation framework. A common strategy is to first identify shared genetic structure at the genome-wide or local level, followed by locus-specific evaluation using joint fine-mapping or colocalization. Additional criteria such as consistency of effect direction and conditional analysis can further refine interpretation, while replication across populations and sensitivity analyses provide robustness checks. Only when multiple lines of evidence converge can shared signals be more confidently interpreted as true pleiotropy (Wang et al., 2024).
3 Genomic Structural Equation Modeling (Genomic SEM)
3.1 Methodological framework
The central aim of Genomic SEM is to explicitly model the shared genetic structure across traits by translating cross-trait covariance into interpretable structural parameters. In this framework, the primary object of inference is not marginal SNP effects for individual traits, but the genetic covariance structure represented by .
The implementation typically follows a two-stage procedure. First, the genetic covariance matrix S is estimated using cross-trait LD Score Regression (LDSC) or related methods, together with a sampling covariance matrix that accounts for sample overlap and other sources of bias. In the second stage, a structural equation model is fitted at the latent level, commonly expressed as:
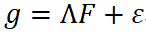
where F represents latent factors capturing shared genetic components, Λ denotes factor loadings, and the residual term ε corresponds to trait-specific effects. Model fitting is usually performed using diagonally weighted least squares (DWLS) or generalized least squares, and model adequacy can be evaluated through standard fit indices such as χ², CFI, and RMSEA (Grotzinger et al., 2019).
At the locus level, Genomic SEM projects SNP-specific effect vectors onto the latent factor space, yielding SNP effects on the shared component, denoted as βFj. In parallel, the QSNP statistic is used to test whether a given SNP deviates from the assumption that its effects are fully mediated through the common factor. It is important to emphasize that QSNP reflects model-based heterogeneity rather than direct evidence of causal mechanisms. Its primary role is to identify loci whose effects are not fully captured by the shared structure, thereby highlighting candidates for further investigation.
Overall, Genomic SEM provides a way to externalize cross-trait genetic relationships into latent structures and to assess the consistency between SNP effects and these structures, rather than to directly infer causal pathways.
3.2 Application scenarios
In studies of comorbid traits, such as obesity, type 2 diabetes, and lipid-related phenotypes, Genomic SEM is often used to construct a latent “metabolic factor” that captures shared genetic influences. Performing GWAS on this latent factor can improve statistical power for detecting variants contributing to shared biological processes. At the same time, SNPs exhibiting significant QSNP values may indicate trait-specific or heterogeneous effects, providing insights into disease subtypes or potential targets for therapeutic intervention.
In psychiatric genetics, Genomic SEM has been applied to model a general liability factor underlying multiple disorders, while preserving disorder-specific components to capture clinical heterogeneity. In plant genetics, similar approaches can be used to model trade-offs among growth, yield, and stress resistance traits, where latent factors may reflect resource allocation or developmental timing. When multi-environment data are available, environmental variables can be incorporated into the model to evaluate gene-environment interactions at the level of shared genetic structure (Grotzinger et al., 2019).
3.3 Strengths and limitations
A major strength of Genomic SEM lies in its ability to model shared genetic structure explicitly at the level of latent variables. Compared to single-trait approaches, it can increase power when genetic effects are consistent across traits, while simultaneously distinguishing shared from trait-specific components through latent factors and heterogeneity statistics. In addition, the explicit modeling of sampling covariance enables the method to account for sample overlap and scale differences, making it suitable for integrative analyses across cohorts.
However, the validity of inference depends critically on the accuracy of the first-stage covariance estimates. Mismatch between LD reference panels and the study population can introduce bias that propagates through the model. As the number of traits increases, model complexity and parameter uncertainty grow substantially, making results sensitive to model specification and identifiability constraints (Grotzinger et al., 2018).
More fundamentally, Genomic SEM characterizes shared genetic structure rather than causal relationships. When the assumed latent structure does not align with the underlying biological mechanisms, the model may attribute correlations arising from linkage disequilibrium or mediation effects to shared pathways. Consequently, results from Genomic SEM should be interpreted in conjunction with locus-level fine-mapping, colocalization analyses, and causal inference methods such as Mendelian randomization.
In this context, locus-level heterogeneity metrics such as QSNP can serve as indicators of complex or non-shared effects, but their interpretation requires validation through downstream analyses and should not be taken as standalone evidence for causality (Figure 1).
.png) Figure 1 Positioning multi-trait structural modeling and locus-level heterogeneity within the causal inference framework
Image caption: This figure illustrates the role of Genomic Structural Equation Modeling (Genomic SEM) in the multi-layered process of causal inference in statistical genetics. Starting from GWAS summary statistics, cross-trait genetic covariance ( |
4 PLEIO: Multi-Trait Joint Fine-Mapping
4.1 Methodological framework
The primary objective of PLEIO is to characterize the structure of shared genetic signals across traits at the locus level. In contrast to single-trait analyses that focus on marginal associations, PLEIO operates on the joint distribution of SNP effects across multiple phenotypes within a genomic region.
The method takes multi-trait GWAS summary statistics as input and models SNP-level effect vectors under local linkage disequilibrium (LD) constraints. Formally, given an LD matrix R and a cross-trait covariance structure Σ, the association statistics for SNP j can be described as:
Zj∼N (0,Σ) (null) or N (μj, Σ) (non-null)
where μj represents the underlying genetic effects across traits, appropriately scaled by sample size and measurement units.
In practice, PLEIO evaluates a set of discrete configurations describing possible patterns of association-such as shared effects across traits, trait-specific effects, or absence of association-within a locus. These configurations are assessed using likelihood-based or Bayesian criteria, often implemented through approximate inference strategies such as expectation-maximization or variational methods. Importantly, these configurations reflect statistical patterns of signal sharing rather than direct evidence of shared causal variants.
To improve robustness, PLEIO explicitly models the cross-trait covariance structure and incorporates intercept terms to account for sample overlap and population structure. The use of structured or sparse priors further stabilizes inference in the presence of weak signals (Hackinger and Zeggini, 2017; Lee et al., 2020; Lee et al., 2021).
Thus, PLEIO provides a locus-level characterization of whether and how genetic signals are shared across traits under LD constraints, serving as an intermediate layer between global structure and causal interpretation.
4.2 Strengths
Joint modeling across traits can substantially increase statistical power, particularly when multiple phenotypes share weak but consistent genetic effects. By leveraging cross-trait covariance, PLEIO effectively aggregates marginal signals, allowing weak associations to accumulate evidence in a multivariate context.
At the locus level, this framework enables more precise resolution of association signals. Compared to single-trait fine-mapping, the integration of multi-trait information can concentrate posterior probabilities on a smaller set of candidate variants, thereby reducing noise induced by LD and improving localization of potential functional variants.
PLEIO is also flexible with respect to trait types, accommodating both quantitative and binary phenotypes, and can incorporate functional annotations to further refine inference. This enables simultaneous integration of genetic architecture across traits and biological priors, facilitating the identification of variants with coherent functional relevance in complex regulatory networks (Lee et al., 2020).
4.3 Limitations
Despite its advantages, PLEIO is sensitive to misspecification of LD structure and cross-trait covariance. When multiple causal variants in high LD affect different traits, the model may incorrectly attribute their effects to a shared signal, thereby inflating false positives. Similarly, inaccuracies in covariance estimation-due to uncorrected sample overlap or measurement heterogeneity-can propagate through the joint likelihood and bias inference.
Differences in phenotype scale (e.g., case-control versus quantitative traits) and strong gene-environment interactions may further complicate effect comparability across traits. In such cases, careful standardization and the use of robust priors (e.g., heavy-tailed or sparse distributions) are necessary to mitigate the influence of outliers.
From a computational perspective, the number of possible effect configurations grows rapidly with the number of traits and candidate variants, leading to increased computational burden and multiple testing concerns. Practical implementations therefore rely on strategies such as genomic windowing, restricting the number of causal variants, or approximate inference methods to balance accuracy and efficiency (Hackinger and Zeggini, 2017; Lee et al., 2020).
More fundamentally, PLEIO identifies patterns of shared association rather than causal relationships. The inferred configurations describe how signals are distributed across traits under LD, but do not establish whether these signals arise from a common causal mechanism. As such, PLEIO results should be integrated with colocalization and causal inference methods, such as Mendelian randomization, to evaluate consistency and potential mediation pathways. Within a broader analytical framework, PLEIO serves as a key locus-level component linking structural modeling (e.g., Genomic SEM) to downstream causal interpretation.
5 Primo: a Multivariate Bayesian Mixture Framework
5.1 Model framework
The core idea of Primo is to represent multi-trait genetic associations as a distribution over discrete patterns rather than as a single shared or trait-specific effect. In this framework, each genetic variant is not simply classified as associated or not, but is assigned probabilities across different combinations of trait associations, thereby providing a probabilistic representation of cross-trait genetic architecture.
Formally, for K traits, each SNP is associated with a binary configuration vector γj ∈{0,1}K, corresponding to 2K possible association patterns. For example, a pattern [1,1,0,… ] indicates that the variant affects the first two traits but not the others. At the level of summary statistics, given the cross-trait covariance matrix Σ, the observed Z-score vector Zj is modeled as:
zj∣γj = p∼N(0,Σ+Vp)
where Vp encodes the effect variance structure associated with pattern p.
Primo introduces prior probabilities πp over all patterns and estimates the posterior probability of each configuration using expectation-maximization, variational inference, or Markov chain methods:
PMPjp = Pr (γj=p∣zj)
Marginal posterior inclusion probabilities (PIPs) for different classes of effects can then be derived by aggregating over pattern subsets, for example:
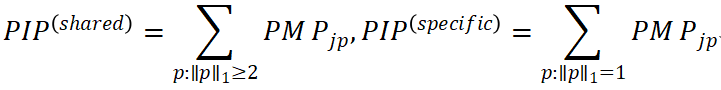
The key feature of this framework is that it does not directly infer causal relationships but instead decomposes cross-trait associations into probabilistic patterns, providing a distributional view of shared and trait-specific effects.
To enhance interpretability, Primo often incorporates hierarchical priors informed by functional annotations (e.g., tissue-specific expression or regulatory features), or introduces structured priors on effect sizes to accommodate concordant or discordant effects across traits (Gleason et al., 2020).
5.2 Applications
In multi-trait settings, Primo provides a shift from analyzing sets of associated loci to characterizing distributions of association patterns. By grouping variants according to their posterior pattern probabilities, genetic signals can be systematically categorized into shared or trait-specific components.
This representation enables downstream enrichment analyses that explicitly condition on pattern type. For example, one can assess whether shared-effect variants are preferentially enriched in specific tissues or regulatory elements, or whether trait-specific signals localize to distinct functional categories. Such analyses offer a basis for generating biologically testable hypotheses.
Posterior pattern probabilities can also serve as a unified scoring metric for prioritizing variants across traits. By distinguishing between “shared-priority” and “trait-specific-priority” sets, and integrating effect direction and functional annotations, it is possible to construct interpretable ranking schemes for candidate loci. In practice, genome-wide screening for high-probability variants is often followed by locus-specific fine-mapping to refine credible sets.
Importantly, these applications operate at the level of statistical pattern characterization and should be interpreted within a broader multi-layered framework that includes structural and locus-level evidence.
5.3 Strengths and limitations
A key strength of Primo lies in its ability to explicitly model cross-trait association patterns. Unlike approaches that dichotomize effects as shared or not, this framework provides a fine-grained probabilistic description of pleiotropic architectures, enabling direct comparison across different patterns within a unified scale.
In addition, the integration of functional annotations within a hierarchical Bayesian framework allows for the combination of statistical evidence with biological priors, improving interpretability and potentially enhancing translational relevance.
However, the method also faces several limitations. The number of possible patterns grows exponentially with the number of traits, leading to substantial computational challenges that require approximate inference or structural constraints. Moreover, inference is sensitive to the specification of cross-trait covariance and LD structure; mismatches in ancestry or incomplete correction for sample overlap may result in spurious sharing signals driven by linkage disequilibrium.
The choice of priors and issues related to parameter identifiability (e.g., label switching) can further affect the stability of posterior estimates, particularly in settings with weak signals or high heterogeneity. For these reasons, Primo is most effective when used in conjunction with complementary approaches, such as structural modeling (e.g., Genomic SEM) and locus-level methods (e.g., PLEIO), to improve robustness.
Most importantly, Primo provides a probabilistic representation of cross-trait association patterns rather than direct evidence of causal relationships. Its outputs should therefore be treated as inputs for downstream analyses, including colocalization and Mendelian randomization, rather than as standalone indicators of shared causal mechanisms (Figure 2).
.png) Figure 2 Probabilistic decomposition of cross-trait association patterns and their network-level interpretation |
6 Network-level Interpretation of Multi-trait Genetic Architecture: from Association Patterns to Functional Structure
6.1 Functional enrichment of cross-trait signals
Within a multi-trait analytical framework, locus-level statistical evidence-whether derived from Genomic SEM (factor effects), PLEIO (joint locus-level signals), or Primo (pattern posterior probabilities)-remains fundamentally a characterization of association structure rather than biological mechanism. Consequently, these signals must be systematically projected onto gene sets, pathways, and higher-order functional structures to enable biologically interpretable insights.
At this stage, the estimand shifts from SNP-level effects to enrichment of signals within functional units. Practically, this requires mapping variants or credible sets to genes using integrative strategies that extend beyond nearest-gene assignment, incorporating eQTL data, chromatin interaction maps, regulatory annotations, and three-dimensional genome architecture. This produces a cross-trait gene-level score matrix that preserves distinctions between shared and trait-specific signals.
Subsequent enrichment analyses (e.g., GSEA, MAGMA, DEPICT) should explicitly correct for LD structure and gene-level confounders such as gene length and SNP density. Critically, shared and trait-specific components should be analyzed separately to distinguish between common biological pathways and trait-divergent processes. Stratified LDSC or annotation-weighted models can further quantify how functional annotations (e.g., tissue specificity, cell type, or developmental timing) differentially contribute to shared versus trait-specific architectures (Pei et al., 2019; Demetci et al., 2021; Pan et al., 2025).
Importantly, inference at this stage constitutes structural interpretation rather than causal inference, describing how genetic signals distribute across functional space without establishing mechanistic directionality.
6.2 Network-based integration of multi-trait signals
Network-based approaches extend beyond gene-set enrichment by incorporating topological relationships among genes. Within protein-protein interaction (PPI), co-expression, transcriptional regulatory, or chromatin interaction networks, signal propagation methods (e.g., random walk or diffusion processes) can aggregate weak effects into coherent modules, thereby mitigating signal fragmentation (Momen et al., 2019; Wu et al., 2020).
Here, the estimand evolves to module-level signal burden and network centrality. Community detection and centrality analyses enable identification of modules or nodes that concentrate cross-trait signals, often representing functional convergence points. In multilayer or multiplex network settings (e.g., across tissues or environmental contexts), higher-order structures linking traits, tissues, and modules can be identified through tensor decomposition or multi-view clustering.
However, network analyses are also susceptible to artifacts introduced at earlier stages. LD structure and population stratification can induce spurious correlations, which may be amplified during network propagation. To mitigate this, permutation-based null models (matched for LD and node degree), multi-ancestry replication, and prior filtering using colocalization evidence are essential (Demetci et al., 2021; Pan et al., 2025). Thus, network-level outputs should be interpreted as organizational patterns of association signals within biological systems, not as direct representations of causal pathways.
6.3 Case study: shared and trade-off architectures in crop traits
In crop genetics, yield and stress resistance traits often exhibit both synergy and trade-offs. Multi-trait analyses can first identify shared and trait-specific loci, which can then be projected onto networks governing root architecture, osmotic regulation, hormonal signaling (e.g., ABA, BR, ethylene), and photosynthetic efficiency. Empirically, under drought or salt-stress conditions, shared signals tend to cluster along a “stress sensing-signal transduction-resource allocation” axis, whereas trade-off loci frequently localize to regulatory branching points between growth and stress response pathways (Momen et al., 2019).
When environmental variation is incorporated, networks can be represented as multilayer structures, with each layer corresponding to a specific condition. Comparing module stability across environments enables the distinction between conserved core modules and context-specific adaptations. From a breeding perspective, prioritizing variants that minimally disrupt network stability may offer a strategy for optimizing yield while maintaining resilience. This perspective reframes breeding as a problem of multi-objective optimization under network constraints, rather than independent trait selection (Pan et al., 2025).
6.4 Case study: network convergence in human metabolic traits
In human complex traits, metabolic phenotypes such as BMI, type 2 diabetes, and lipid profiles exhibit substantial genetic overlap. Mapping shared and trait-specific signals onto cross-tissue regulatory networks (e.g., liver, adipose, muscle) reveals convergence in pathways related to insulin signaling, lipid metabolism, and inflammation (Pei et al., 2019).
At the network level, shared signals often concentrate in central regulatory modules, whereas trait-specific signals localize to peripheral pathways. For example, T2D-specific signals are enriched in pancreatic β-cell function, while lipid-specific signals are associated with lipoprotein metabolism.
Further integration with causal inference tools (e.g., MR or mediation analysis at the module level) allows testing of directional hypotheses, such as inflammatory pathways contributing to insulin resistance and downstream metabolic dysregulation (Wu et al., 2020). Overlaying drug targets onto these networks enables prioritization strategies based on both module membership and cross-tissue centrality, potentially improving therapeutic efficacy while minimizing off-target effects (Demetci et al., 2021).
6.5 Summary: role and limits of the network layer
In summary, network-level analysis serves to organize locus- and pattern-level evidence into a structured functional landscape. The key estimands at this level include: enrichment of signals within functional categories; module-level cross-trait signal aggregation; topological importance of network nodes.
However, these remain structural descriptors rather than causal estimates. Their primary value lies in identifying candidate shared biological mechanisms; guiding downstream causal inference (e.g., colocalization and MR); prioritizing targets for experimental validation and translation. Accordingly, the network layer should be viewed as an intermediate bridge linking statistical association patterns to causal mechanisms, rather than as a final inferential endpoint.
7 Bias-variance Trade-offs and Method Selection: a Risk-function Perspective in Multivariate Genetic Inference
7.1 A unified simulation framework aligned with estimands
In multi-trait genetic analysis, methodological differences do not arise solely from implementation details, but fundamentally reflect differences in their underlying estimands. Genomic SEM targets shared latent genetic factors at the structural level, PLEIO focuses on locus-level joint effects and causal configurations, whereas Primo characterizes the probabilistic distribution of cross-trait association patterns. Consequently, method evaluation should be framed not merely in terms of detection power, but in relation to the bias-variance properties associated with each estimand.
To enable such evaluation, a unified simulation framework can be constructed by systematically varying key parameters, including genetic correlation (rg), LD structure (single- vs. multi-peak, simple vs. complex, matched vs. mismatched ancestry), causal variant density, effect direction (concordant, discordant, mixed), sample size and overlap proportion, as well as error distribution and measurement noise. Under simulated or empirically derived genotype data, multi-trait GWAS summary statistics can be generated and analyzed across methods, allowing comparison under consistent metrics such as bias, variance, mean squared error (MSE), confidence interval coverage, and false discovery control.
Within this framework, a consistent bias-variance trade-off emerges. More flexible models (e.g., high-dimensional SEM or pattern-based approaches such as Primo) are better able to capture complex genetic architectures, thereby reducing structural bias, but often at the cost of increased variance under limited sample size or complex LD. Conversely, more constrained models tend to exhibit lower variance but may incur higher bias when their assumptions are violated (Liu and Rhemtulla, 2021; Ranglani, 2024). Thus, method performance should be interpreted as a balance between model adequacy and estimation stability.
7.2 Method behavior under heterogeneous genetic architectures
When substantial shared genetic structure exists across traits (moderate to high rg, concordant effect directions), Genomic SEM and PLEIO can effectively aggregate signals, improving power and reducing estimation variance. However, such gains depend critically on the validity of the shared-structure assumption. When the true architecture deviates (e.g., presence of opposing effects or multiple causal variants), model misspecification can introduce substantial bias.
In sparse-effect settings with a mixture of shared and trait-specific signals, Primo tends to provide more robust control of false discoveries by explicitly modeling association patterns, albeit sometimes at the expense of reduced sensitivity. This advantage becomes particularly evident when distinct causal variants reside in strong LD, where locus-level joint models may incorrectly infer shared causality.
Under conditions of pronounced heterogeneity-such as opposing effect directions or ancestry-dependent LD differences-the common-factor assumption in Genomic SEM may be violated. In such cases, heterogeneity statistics (e.g., QSNP) provide critical diagnostic signals, indicating the need for complementary locus-level or external analyses. Similarly, sample overlap and LD misspecification can systematically inflate variance and type I error rates across all methods, necessitating intercept correction and sensitivity analyses. Overall, differences in method performance reflect the boundaries of their respective estimands, rather than inherent superiority of one method over another.
7.3 Empirical validation: linking statistical performance to biological consistency
In real data applications, method evaluation must consider both statistical performance and biological interpretability. In crop systems, multi-environment phenotypic data (e.g., yield, flowering time, stress tolerance) provide a suitable testbed. After identifying candidate regions via genome-wide and local genetic correlation, PLEIO and Primo can be compared in terms of shared signal identification and credible set refinement, with replication across environments and functional annotations serving as external validation. Genomic SEM can then be used to construct latent factors and perform factor-based GWAS, allowing assessment of concordance between structural- and locus-level inference.
In human complex traits (e.g., metabolic syndrome phenotypes), multi-trait models often reveal strong shared genetic components. In this context, Genomic SEM enhances power at the pathway level, PLEIO improves locus resolution, and Primo enables classification of shared versus trait-specific patterns. Integration with eQTL colocalization, drug target annotations, and cross-ancestry replication further supports biological validation (Chen et al., 2023). Such dual evaluation-statistical performance alongside biological consistency-helps mitigate over-reliance on statistical significance alone.
7.4 Principles for method selection: an estimand-driven framework
Method selection should be guided by the target estimand rather than heuristic preference: when the goal is to identify shared genetic architecture or common pathways, Genomic SEM and PLEIO are generally preferable; when distinguishing shared versus trait-specific effects under heterogeneity, Primo offers a more direct probabilistic framework; under complex or heterogeneous scenarios, methods with weaker structural assumptions should be prioritized, complemented by cross-method validation.
In practice, a staged analysis strategy is recommended. First, use genetic correlation to identify sets of traits suitable for joint analysis. Second, apply PLEIO and Primo at candidate loci to cross-validate shared versus specific effects. Finally, perform genome-wide expansion using Genomic SEM on confirmed shared structures. This layered approach enables systematic progression from structure identification to signal amplification.
7.5 Reporting standards and reproducibility
To ensure interpretability and reproducibility, the following elements should be explicitly reported: definitions and applicability of each method’s estimand; bias, variance, and MSE under the chosen analytical framework; credible set size and cross-ancestry consistency; treatment of sample overlap and LD reference, including sensitivity analyses; evidence grading for true versus spurious pleiotropy. Full transparency in analytical pipelines, parameter settings, and code availability is essential to support reproducibility and robust comparison across studies (Chen et al., 2023; Ranglani, 2024).
8 Discussion: Hierarchical Integration of Multi-trait Inference and Methodological Boundaries
The principal advantage of multi-trait analysis lies in its ability to integrate information through the covariance structure among traits, thereby establishing a closer connection between statistical power and biological interpretation. However, this “borrowing of strength” is not unconditional. Its validity depends on the correct alignment of estimands across inferential layers and on a strict distinction between their respective interpretive boundaries.
At the structural level, when traits exhibit at least moderate genetic correlation (e.g., rg>0.2) and largely concordant effect directions, Genomic SEM can aggregate signals at the genome-wide level through latent factor modeling, thereby improving detection power and revealing shared genetic architecture. At the locus level, PLEIO integrates cross-trait evidence within local LD windows through joint modeling, which helps refine credible sets and enhances the detectability of weak-effect loci. At the pattern level, Primo decomposes shared-versus-specific architectures through posterior probabilities, reorganizing dispersed weak signals into interpretable probabilistic patterns (Turley et al., 2018). These three approaches are therefore complementary across layers, corresponding respectively to structural, locus-level, and pattern-level estimands.
The value of this hierarchical framework has already been demonstrated in practice. In crop genetic improvement, multi-trait models that jointly analyze yield and stress-resistance traits can improve predictive accuracy and optimize selection strategies (Velazco et al., 2019; Bhatta et al., 2020). In human genetics, multi-trait joint analysis has substantially increased the number of detectable loci and improved the interpretability of polygenic risk scores (Turley et al., 2018). Together, these findings indicate that multivariate frameworks are more sensitive to shared genetic bases and are capable of revealing structural convergence at the pathway or network level.
At the same time, multi-trait analysis also amplifies inferential risk, particularly with respect to the problem of spurious pleiotropy. Variants that are tightly linked but causally distinct, ancestry-related LD mismatch, sample overlap, and measurement error may all cause apparent correlation to be misinterpreted as shared causality. Under multi-trait settings, these factors may be magnified through the covariance structure, thereby affecting inference at all downstream layers. In addition, methodological complexity increases rapidly with the number of traits: the identifiability of Genomic SEM, the configuration search burden of PLEIO, and the exponentially expanding pattern space of Primo all pose challenges for computational efficiency and parameter stability (Lozano et al., 2023).
More importantly, differences in scale and distribution across traits-such as binary versus quantitative phenotypes or the presence of strong gene-by-environment (G×E) effects-can directly influence covariance estimation and effect comparability, thereby affecting all subsequent analyses. The reliability of multi-trait inference therefore depends not only on the methods themselves, but also on the consistency and quality control of the input data.
Looking ahead, a key direction for future development is the transition from “single-layer evidence” to “cross-layer consistency.” First, at the covariance level, methods capable of capturing nonlinear or hierarchical correlation structures should be further developed, ideally integrating both genome-wide and local genetic correlation information to improve structural inference. Second, at the locus level, joint fine-mapping and colocalization should become routine components of analysis, with overlap of credible sets and shared posterior probabilities used to evaluate the robustness of shared causality. Third, at the model level, sample overlap, LD reference choice, and latent variable specification should be incorporated into a systematic sensitivity-analysis framework. Furthermore, in multi-ancestry and multi-omics settings, cross-ancestry replication and integration of functional annotations will be essential to avoid misclassifying mediated effects as horizontal pleiotropy. Finally, at the causal level, Mendelian randomization and mediation analysis should be embedded within the multi-trait framework to distinguish shared causality from chain-mediated causal effects, thereby establishing operational criteria for inference.
At the application level, although crop and human systems differ in data structure and research objectives, their paths of methodological integration show notable convergence. In crop breeding, multi-trait frameworks help dissect the trade-off between productivity and resilience: Genomic SEM identifies common factors underlying resource allocation, PLEIO localizes candidate loci within key pathways, and Primo distinguishes shared from trait-specific architectures, thereby supporting network-constrained multi-objective selection strategies (Velazco et al., 2019; Bhatta et al., 2020; Mbebi et al., 2025). In human medicine, a similar framework can be applied to metabolic syndrome or psychiatric disease spectra: factor-based GWAS reveals cross-trait susceptibility, PLEIO improves locus-level resolution, and Primo provides pattern classification and integration with functional annotation, thereby informing the prioritization of candidate drug targets (Turley et al., 2018).
In summary, multi-trait analysis is not merely a replacement for single methods, but rather a cross-layer inferential system. Its effective application depends on explicitly defining the estimand targeted by each method and establishing a coherent chain of evidence across the structural, locus, pattern, and causal layers. At the same time, standardized robustness evaluation and cross-ancestry validation remain essential prerequisites for improving reproducibility and generalizability.
9 Conclusion
In this study, multi-trait statistical genetics is reframed from a collection of parallel methods into a unified inference framework driven by clearly defined estimands. Within this framework, Genomic SEM, PLEIO, and Primo are not interchangeable tools, but operate on distinct inferential layers: Genomic SEM characterizes the genetic covariance structure and latent shared factors across traits, whereas PLEIO and Primo resolve cross-trait effects at the locus and pattern levels, respectively. Together, these approaches form a hierarchical system spanning covariance (structure) → locus configuration → association pattern, enabling a progressive refinement of evidence rather than isolated hypothesis testing.
Under this perspective, the value of multi-trait analysis extends beyond gains in statistical power. It lies in the ability to decompose genetic architecture into interpretable components: latent factor modeling aggregates weak but coherent signals, while locus- and pattern-level inference distinguishes shared from trait-specific effects. This “detection-decomposition-reconstruction” process transforms dispersed associations into biologically structured units, facilitating downstream interpretation at the pathway and network levels and supporting translational applications in both crop improvement and disease biology.
A central requirement for reliable inference is the rigorous distinction between true and spurious pleiotropy. Apparent cross-trait signals may arise not only from shared causal variants, but also from linkage disequilibrium, sample structure, or mediated relationships. We therefore emphasize a multi-evidence workflow integrating local genetic correlation, joint fine-mapping and colocalization, effect direction consistency, and cross-ancestry or family-based validation, accompanied by systematic sensitivity analyses and false discovery rate control. Such integration is essential to constrain inference both statistically and biologically.
In practice, we advocate a structured “screen-validate-apply” workflow. First, genome-wide and local genetic correlation analyses identify trait sets with shared genetic backgrounds. Second, candidate regions are interrogated using complementary approaches such as PLEIO and Primo, together with conditional analysis and colocalization, to resolve shared versus specific effects. Finally, confirmed shared structures are expanded genome-wide via Genomic SEM factor-based analysis and integrated into network enrichment and functional interpretation pipelines, supporting breeding strategies or therapeutic prioritization.
Overall, the strength of multi-trait frameworks does not reside in joint analysis per se, but in maintaining alignment between estimands and interpretation across inferential layers. When accompanied by transparent reporting of bias-variance trade-offs, LD and ancestry matching, and sample structure, multi-trait approaches can evolve from tools of association detection into integrative systems linking genetic architecture, molecular mechanisms, and actionable biological insights. This transition marks a critical step from descriptive correlation toward mechanistic understanding in complex trait genetics.
Author Contributions
Xuanjun Fang conducted this study, including literature review, data analysis, and the writing and revision of the manuscript. The author has read and approved the final version of the manuscript.
Acknowledgements
This work was supported by a Major Project of the National Natural Science Foundation of China (Grant No. 30490254).
Bhatta M., Gutiérrez L., Cammarota L., Cardozo F., Germán S., Gómez-Guerrero B., Pardo M., Lanaro V., Sayas M., and Castro A., 2020, Multi-trait genomic prediction model increased the predictive ability for agronomic and malting quality traits in barley (Hordeum vulgare L.), G3: Genes, Genomes, Genetics, 10(3): 1113-1124.
https://doi.org/10.1534/g3.119.400968
Chen Z., Zhang J., Sarro F., and Harman M., 2023, A comprehensive empirical study of bias mitigation methods for machine learning classifiers, ACM Transactions on Software Engineering and Methodology, 32(4): 1-30.
https://doi.org/10.1145/3583561
Demetci P., Cheng W., Darnell G., Zhou X., Ramachandran S., and Crawford L., 2021, Multi-scale inference of genetic trait architecture using biologically annotated neural networks, PLoS genetics, 17(8): e1009754.
https://doi.org/10.1101/2020.07.02.184465
Fang X.J., and Wu W.R., 2026, Evolution of statistical genetic paradigms: from linkage analysis and candidate gene strategies to GWAS, Molecular Plant Breeding, 24(9): 2817-2829.
Gleason K., Yang F., Pierce B., He X., and Chen L., 2020, Primo: Integration of multiple GWAS and omics QTL summary statistics for elucidation of molecular mechanisms of trait-associated SNPs and detection of pleiotropy in complex traits, Genome Biology, 21(1): 236.
https://doi.org/10.1186/s13059-020-02125-w
Grotzinger A.D., Rhemtulla M., de Vlaming R., Ritchie S.J., Mallard T.T., Hill W.D., Ip H.F., Marioni R.E., McIntosh A.M., Deary I.J., Koellinger P.D., Harden K.P., Nivard M.G., and Tucker-Drob E.M., 2018, Genomic SEM provides insights into the multivariate genetic architecture of complex traits, bioRxiv, 2018: 305029.
https://doi.org/10.1101/305029
Grotzinger A.D., Rhemtulla M., de Vlaming R., Ritchie S.J., Mallard T.T., Hill W.D., Ip H.F., Marioni R.E., McIntosh A.M., Deary I.J., Koellinger P.D., Harden K.P., Nivard M.G., and Tucker-Drob E.M., 2019, Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits, Nature human behaviour, 3(5): 513-525.
https://doi.org/10.1038/s41562-019-0566-x
Hackinger S., and Zeggini E., 2017, Statistical methods to detect pleiotropy in human complex traits, Open Biology, 7(11): 170125.
https://doi.org/10.1098/rsob.170125
Lee C., Shi H., Pasaniuc B., Eskin E., and Han B., 2020, A method to map and interpret pleiotropic loci using summary statistics of multiple traits, bioRxiv, 2020: 155879.
https://doi.org/10.1101/2020.06.16.155879
Lee C., Shi H., Pasaniuc B., Eskin E., and Han B., 2021, PLEIO: A method to map and interpret pleiotropic loci with GWAS summary statistics, The American Journal of Human Genetics, 108(1), 36-48.
https://doi.org/10.1016/j.ajhg.2020.11.017
Liu S., and Rhemtulla M., 2021, Treating random effects as observed versus latent predictors: The bias-variance tradeoff in small samples, British Journal of Mathematical and Statistical Psychology, 75(1): 158-181.
https://doi.org/10.1111/bmsp.12253
Lozano A., Ding H., Abe N., and Lipka A., 2023, Regularized multi-trait multi-locus linear mixed models for genome-wide association studies and genomic selection in crops, BMC Bioinformatics, 24(1): 399.
https://doi.org/10.1186/s12859-023-05519-2
Mbebi A., Mercado F., Hobby D., Tong H., and Nikoloski Z., 2025, Advances in multi-trait genomic prediction approaches: classification, comparative analysis, and perspectives, Briefings in Bioinformatics, 26(3): bbaf211.
https://doi.org/10.1093/bib/bbaf211
Momen M., Campbell M., Walia H., and Morota G., 2019, Utilizing trait networks and structural equation models as tools to interpret multi-trait genome-wide association studies, Plant Methods, 15(1): 107.
https://doi.org/10.1186/s13007-019-0493-x
Pan Q., Bauters M., Peaucelle M., Ellsworth D., Kattge J., and Verbeeck H., 2025, Network-informed analysis of a multivariate trait-space reveals optimal trait selection, Communications Biology, 8(1): 569.
https://doi.org/10.1038/s42003-025-07940-0
Pei G., Sun H., Dai Y., Liu X., Zhao Z., and Jia P., 2019, Investigation of multi-trait associations using pathway-based analysis of GWAS summary statistics, BMC Genomics, 20(Suppl 1): 79.
https://doi.org/10.1186/s12864-018-5373-7
Ranglani H., 2024, Empirical analysis of the bias-variance tradeoff across machine learning models, Machine Learning and Applications: An International Journal, 11(4): 1-15.
https://doi.org/10.5121/mlaij.2024.11401
Turley P., Walters R.K., Maghzian O., Okbay A., Lee J.J., Fontana M.A., Nguyen-Viet T.A., Wedow R., Zacher M., Furlotte N.A., 23andMe Research Team, Social Science Genetic Association Consortium, Magnusson P., Oskarsson S., Johannesson M., Visscher P.M., Laibson D., Cesarini D., Neale B.M., and Benjamin D.J., 2018, Multi-trait analysis of genome-wide association summary statistics using MTAG, Nature Genetics, 50(2): 229-237.
https://doi.org/10.1038/s41588-017-0009-4
Velazco J.G., Jordan D.R., Mace E.S., Hunt C.H., Malosetti M., and van Eeuwijk F.A., 2019, Genomic prediction of grain yield and drought-adaptation capacity in sorghum is enhanced by multi-trait analysis, Frontiers in Plant Science, 10: 997.
https://doi.org/10.3389/fpls.2019.00997
Wang X., Wang J., Xia X., Xu X., Li L., Cao S., Hao Y., and Zhang L., 2024, Effect of genotyping errors on linkage map construction based on repeated chip analysis of two recombinant inbred line populations in wheat (Triticum aestivum L.), BMC Plant Biology, 24(1): 306.
https://doi.org/10.1186/s12870-024-05005-8
Wu Y., Cao H., Baranova A., Huang H., Li S., Cai L., Rao S., Dai M., Xie M., Dou Y., Hao Q., Zhu L., Zhang X., Yao Y., Zhang F., Xu M., and Wang Q., 2020, Multi-trait analysis for genome-wide association study of five psychiatric disorders, Translational Psychiatry, 10(1): 209.
https://doi.org/10.1038/s41398-020-00902-6
Zhang L., Li H., and Wang J., 2015, Linkage analysis and map construction in genetic populations of clonal F1 and double cross, G3: Genes, Genomes, Genetics, 5(3): 427-439.
https://doi.org/10.1534/g3.114.016022




. HTML
Associated material
. Readers' comments
Other articles by authors
. Xuanjun Fang
Related articles
. Multi-trait genetic analysis
. Pleiotropy
. Genetic correlation (rg)
. Genomic SEM
. Joint fine-mapping
. Bayesian mixture models
. Colocalization
. Causal inference
Tools
. Post a comment